
Mitochondrial diseases are a complex group of disorders, with many different symptoms and many different genetic causes. Mitochondrial diseases are known as ‘mito’ for short.
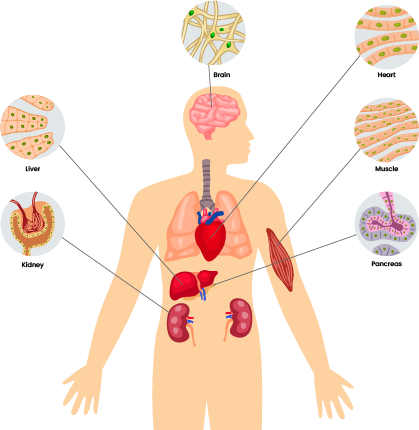
'Mitochondrial diseases' is a collective term for a group of disorders that particularly affect the brain, heart, liver, skeletal muscles, kidney and the endocrine and respiratory systems. There are many different types of mitochondrial diseases (see Types).
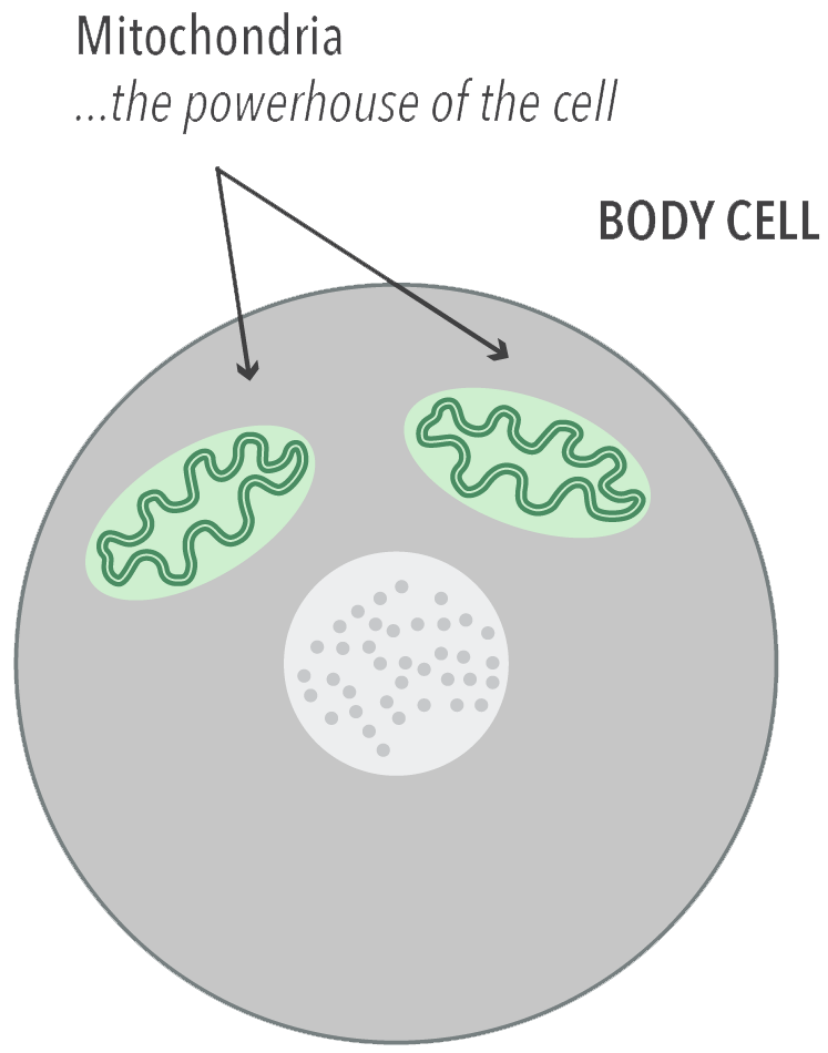
Our bodies are made up of different tissues, for example muscle, nerve, and liver. Each tissue is composed of small 'building blocks', called cells, and within each cell are mitochondria. The job of the mitochondria is to generate energy. Just like a power station, they take in fuel (the food we eat) and use it to generate energy. If this process fails, the cell cannot function adequately and this can lead to disease. Muscle and brain require a lot of energy and are often the most severely affected.
The part of mitochondria involved in energy production is called the respiratory chain. Components (called proteins) of this respiratory chain pathway are produced from genetic instructions (the DNA) found either within the mitochondria themselves (the mitochondrial DNA, or mtDNA), or in the nuclear DNA (nDNA) that is found within the nucleus of the cell.
Mito occurs when the mitochondria aren’t working properly. Mitochondria are responsible for creating more than 90% of the energy needed by the body to sustain life and support growth. When they fail, less and less energy is generated within the cell. Mito most often affects the parts of the body which need a lot of energy: the brain, heart, liver, skeletal muscles, kidney and the endocrine and respiratory systems.
Depending on which cells are affected, symptoms may include loss of motor control, muscle weakness and pain, gastro-intestinal disorders and swallowing difficulties, poor growth, cardiac disease, liver disease, diabetes, respiratory complications, seizures, visual/hearing problems, lactic acidosis, developmental delays and susceptibility to infection. If the process of cell failure is repeated throughout the body, whole systems begin to fail, and quality of life can be severely impacted.

Are mitochondrial diseases hereditary?
Like everything about mito, this is complex. Some mitochondrial disorders, even though they involve the mitochondrial DNA, happen spontaneously. This means that only one individual in a family is affected - the parents and any children of that person are unaffected. Other mitochondrial disorders are only inherited from the mother – these are known as maternally inherited mitochondrial diseases. Disorders that arise because of a change (mutation) within the genes found within the nucleus may be inherited from either or both parents. Often parents are unaware that they carry a genetic change that can lead to their child developing mito and it is only once the child is diagnosed that the parent(s) is also diagnosed.
Energy factories and much more
The conventional teaching in biology and medicine is that mitochondria function only as "energy factories" for the cell. This over-simplification is a mistake which has slowed our progress toward understanding the biology underlying mitochondrial disease. It takes about 3000 genes to make a mitochondrion. Mitochondrial DNA encodes just 37 of these genes; the remaining genes are encoded in the cell nucleus and the resultant proteins are transported to the mitochondria. Only about 3% of the genes necessary to make a mitochondrion (100 of the 3000) are allocated for making ATP. More than 95% (2900 of 3000) are involved with other functions tied to the specialized duties of the differentiated cell in which it resides.
These duties change as we develop from embryo to adult, and our tissues grow, mature, and adapt to the postnatal environment. These other, non-ATP-related functions are intimately involved with most of the major metabolic pathways used by a cell to build, break down, and recycle its molecular building blocks. Cells cannot even make the RNA and DNA they need to grow and function without mitochondria. The building blocks of RNA and DNA are purines and pyrimidines. Mitochondria contain the rate-limiting enzymes for pyrimidine biosynthesis (dihydroorotate dehydrogenase) and heme synthesis (d-amino levulinic acid synthetase) required to make hemoglobin. In the liver, mitochondria are specialized to detoxify ammonia in the urea cycle. Mitochondria are also required for cholesterol metabolism, for estrogen and testosterone synthesis, for neurotransmitter metabolism, and for free radical production and detoxification. They do all this in addition to breaking down (oxidizing) the fat, protein, and carbohydrates we eat and drink.
Defining mitochondrial disease

Mito is the result of either inherited or spontaneous mutations in mtDNA or nDNA which prevent the mitochondria from working properly. The condition can affect anyone, at any age and symptoms may be seen in only one tissue or can occur throughout the body. This often means that mito can affect people in very different ways. More research is needed to help us fully understand these differences.
The complexity of mitochondrial disease
Because mitochondria perform so many different roles within the cell, there are literally hundreds of different mitochondrial disorders. Each disorder produces a spectrum of symptoms that can be confusing to both patients and physicians in early stages of diagnosis. Because of the complex interplay between the hundreds of genes and cells that must cooperate to keep our metabolic machinery running smoothly, it is a hallmark of mitochondrial diseases that identical mtDNA mutations may not produce identical diseases.
The opposite can also happen where different mutations in mtDNA and nDNA can lead to the same diseases. An example of this is Leigh syndrome, which can be caused by about a dozen different gene defects.
Leigh syndrome may be caused by the NARP mutation, the MERRF mutation, complex I deficiency, cytochrome oxidase (COX) deficiency, pyruvate dehydrogenase (PDH) deficiency, and other unmapped DNA changes. Not all patients with these DNA abnormalities will go on to develop Leigh syndrome, however.
Mitochondrial diseases are even more complex in adults because detectable changes in mtDNA occur as we age and, conversely, the aging process itself may result from reduced mitochondrial function. Abnormal mitochondrial function has been seen in a broad spectrum of metabolic, inherited and acquired disorders in adults.
Mito can be so complex that even doctors and researchers find it difficult to understand – so it’s unsurprising that people affected are often overwhelmed and confused. These feelings are very normal, particularly when first diagnosed or when a new symptom appears.

Denis Archibald Leigh